



L’arbre du vivant, une construction perpétuelle
On sait que toute la vie sur Terre est unie par des liens de parenté universels. Mais comment les scientifiques sont-ils parvenus à élaborer une classification ?
Dans un précédent article était évoqué le sens complexe du mot “poisson” dans la biologie moderne. Par une définition phylogénétiquement stricte, nous, humains, et tous les autres vertébrés terrestres également, sommes des poissons, car nous sommes leurs descendants. Nous sommes d’ailleurs plus proches parents du saumon que du requin, et plus proches du coelacanthe que du saumon ! L’arbre ci-dessous illustre ce schéma de parenté. En effet, nous descendons d'un cousin des poissons osseux actuels (comme le saumon), et non cartilagineux (comme le requin), et d'un cousin des poissons à nageoires lobées (comme le cœlacanthe) et non rayonnantes (comme le saumon). Ainsi, les humains partagent avec le saumon des liens de parenté plus étroits que chacun des deux ne partage avec le requin - nous partageons un ancêtre commun plus récent (qui aurait été un poisson osseux). Et cela de même que nous sommes plus apparentés au gorille qu’au macaque (avec comme ancêtre commun un grand singe sans queue), ou que l’on est plus apparenté à son frère qu’à sa cousine (avec comme ancêtres communs deux parents, plus récents que deux grands-parents). Le même raisonnement fait de nous et du cœlacanthe des cousins proches, par rapport au saumon plus éloigné - un ancêtre commun à nageoires lobées plus récent nous unit, par rapport à notre ancêtre commun à tous les trois (dont la forme des nageoires n’est pas connue avec certitude). Si, à l’aide de l’arbre ci-dessous, vous avez pu suivre ce raisonnement, vous possédez les bases pour lire un arbre phylogénétique (voir le glossaire à la fin de l’article pour un rappel précis des termes clés). Mais comment en sommes-nous arrivés là, scientifiquement parlant ?
Le mot “poisson”, à l'exclusion des vertébrés terrestres et de leurs descendants, est un “grade” ou groupe paraphylétique en termes cladistiques. Ce n'est pas un groupe “naturel” ou monophylétique, incluant tous les descendants d'un même ancêtre commun. Dans cet arbre phylogénétique des vertébrés, les bouts de branches sont des espèces ou taxons dont on cherche à illustrer la parenté, les nœuds sont des ancêtres communs entre ces taxons, et les branches représentent une lignée évolutive théorique d’un ancêtre vers ses descendants. Ces ancêtres et lignées sont théoriques, dans la mesure où ils sont déduits par l’étude des taxons “descendants”.
De la scala naturae à l’arbre moderne
Depuis l'antiquité, ceux qui se plongent dans l'étude de la nature cherchent à comprendre la structure du vivant. On a longtemps placé les organismes dans une chaîne hiérarchique, la scala naturae (littéralement “échelle naturelle” en latin) avec l'homme tout en haut. La combinaison de cette idée de progression du vivant, avec le schéma diagrammatique et métaphorique de l’arbre (notamment utilisé en généalogie), a donné naissance à l’idée d'un arbre du vivant, avant même que les idées de descendance commune universelle et d’évolution ne voient le jour. La remise en cause progressive de cette conception hiérarchisée s’est accompagnée de la formalisation de la taxonomie (classification) démarrée par Carl Von Linné, et s’est cristallisée avec l’avènement de la biologie moderne. Les travaux de Charles Darwin, de ses contemporains, et des grands naturalistes qui le précédaient, ainsi que la “synthèse évolutive inclusive” qui a émergé plus de 150 ans après L'Origine des espèces (grâce aux apports de la génétique, l’écologie, la biologie évolutive moderne, etc.) ont définitivement enterré cette conception. L’évolution est constante pour toutes les espèces, et elle leur offre la possibilité de s’adapter de mieux en mieux, mais spécifiquement à leur environnement, pas dans l’absolu. L’arbre du vivant moderne, l’arbre phylogénétique, sert donc à illustrer les liens de parenté entre espèces, sans hiérarchie implicite. Ou plutôt, comme on verra, il représente une hypothèse de schéma de parenté entre espèces, sur la base des données biologiques et des outils analytiques et mathématiques utilisés.
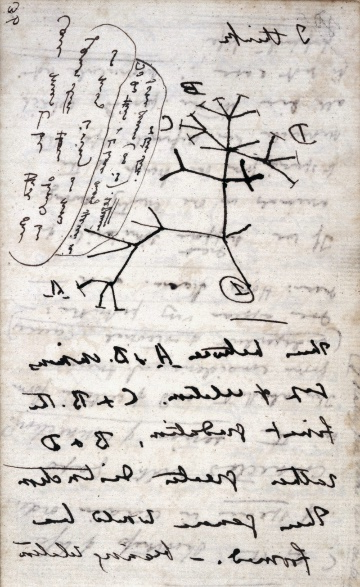
Déjà dans ses notes préparatoires pour son œuvre majeure, Darwin fait usage d'un schéma en forme d'arbre pour figurer les liens de parenté entre organismes. A gauche, l'arbre présent dans L'Origine des espèces.
Le travail des zoologistes, botanistes, mycologistes et microbiologistes, qui s’attaquent encore aujourd'hui à comprendre l'arbre du vivant, est donc l’étude de la systématique (ou phylogénie). C'est Willi Hennig, un entomologiste allemand qui, au milieu du XXe siècle, en formalisant la notion de parenté entre espèces, a fixé le socle de la pensée systématique moderne : l’identification de groupes monophylétiques, c'est-à-dire incluant tous les descendants d’un même ancêtre commun sans exception. Par exemple, le groupe monophylétique des dinosaures inclut les descendants actuels de certains dinosaures théropodes, tels les oiseaux. Le terme “poisson”, au sens monophylétique, inclurait tous les vertébrés. Dans la cladistique (que Hennig appelait “systématique phylogénétique”), ces groupes, appelés clades, sont identifiés à l’aide de caractères dérivés qui leur sont uniques, dits synapomorphiques. Hennig se base sur le concept historique d'homologie biologique, c'est-à-dire sur la similarité des structures morphologiques liée à la parenté sous-jacente (à noter la correspondance de structures entre le bras d’un humain, la patte d'un chat, l'aile d’une chauve-souris). Ici, il l’applique spécifiquement dans le contexte de caractères dérivés (non ancestraux) et qui sont ainsi diagnostiqués d’un clade particulier, relatifs à un groupe apparenté voisin. Par exemple, la possession de quatre membres chez les tétrapodes, contrairement aux nageoires en nombre varié des poissons lobés qui leur étaient apparentés, représente une synapomorphie du groupe. En identifiant des caractères potentiellement synapomorphiques, on collecte les données nécessaires à la génération d'un arbre phylogénétique.
Des branches ou des boîtes ?
Mais cartographier le vivant participe aussi implicitement à établir sa classification, donc à faire un travail de taxinomie (ou taxonomie). La taxonomie reste pleinement à l'ordre du jour tant elle nous est nécessaire pour comprendre intuitivement la structure du vivant. Cependant il convient de rappeler que les catégories que nous établissons, même sur une base phylogénétique, restent des étiquettes quelque peu artificielles. La taxonomie est une schématisation subjective de la vie, simplification qui nous permet d’appréhender la biodiversité telle qu'elle existe actuellement ou a existé par le passé, et de l’étudier. Le vivant est un continuum d'individus apparentés entre eux, donc un réseau sur la base duquel on peut générer un nombre incalculable de catégories emboîtées. Et sur le temps long géologique, ces catégories tendent à perdre leur sens. En effet, tous les oiseaux modernes descendent d'une seule et unique espèce de dinosaure avien, et tous les mammifères modernes descendent d'une seule espèce de “reptile” synapside. Impossible en pratique de délimiter rigoureusement le clade “espèce de l'ancêtre commun” et le clade “classe de vertébrés” (mammifères et oiseaux correspondent à des classes en taxonomie). Et on pourrait écrire un article entier sur la nature problématique du mot “espèce”. La phylogénie, ou l'élucidation de l’arbre du vivant - du réseau de parenté - reste donc la réelle tâche fondamentale du travail d'histoire naturelle.
Les caractères : les graines de l’arbre phylogénétique
La cladistique de l'époque d'Hennig se reposait sur les données historiquement utilisées pour étudier l'évolution : les données morphologiques, provenant de l'étude de l'anatomie des organismes. C'est sur la base de la morphologie que les chercheurs identifiaient des groupes taxonomiques, et la méthode cladistique permettait d'apporter de la rigueur mathématique à ce processus. Le travail de la cladistique commence donc par lister différents caractères morphologiques d'un échantillon d'organismes, avec un état par caractère pour chaque organisme (par exemple nageoire rayonnante ou lobée), et un coût de transition entre chacun des états d’un caractère (passer de 4 à 5 doigts n’est pas équivalent à passer d’une main à une aile, en terme de “distance évolutive”) - transition évolutive qui aurait lieu dans les branches de l’arbre, entre les nœuds représentant les organismes ou leurs ancêtres. A partir de là, les systématistes peuvent quantifier la “distance phylogénétique” entre deux espèces, ou donner à chaque arbre potentiel un score de nombre de transitions nécessaires, ou de vraisemblance statistique. De premiers arbres phylogénétiques ont ainsi pu être produits à la main, même si l'apparition de l’outil informatique a rapidement été privilégié au vu de la complexité exponentielle de la tâche - avec la multiplication des espèces ou caractères inclus dans un arbre.
Au cours de ce même XXe siècle, et notamment dans sa seconde moitié, l’étude de la génétique était en pleine expansion. Avec l'arrivée des techniques de séquençage et leur perfectionnement progressif, les données moléculaires (génétiques ou protéiques) sont devenues disponibles à la systématique. Cela a notamment offert, pour la première fois, la possibilité de faire des phylogénies d'organismes unicellulaires, aux caractères morphologiques relativement limités et cryptiques. Les premières phylogénies moléculaires étaient basées sur des protéines, avant que L'ADN et l'ARN soient aussi implémentés. Dans une séquence moléculaire, chaque position dans la séquence constitue un caractère, avec 4 états (les nucléotides ATGC pour l'ADN ou AUGC pour l'ARN) ou 22 états (les acides aminés pour les protéines) possibles. Les séquences orthologues (gènes homologues entre espèces) doivent être alignées au préalable, en prenant en compte que certaines séquences auront des positions vides (perte d'une partie de séquence). Un modèle de substitution est nécessaire pour donner un coût à chaque substitution possible entre nucléotides, ou entre acides aminés (on n’estime pas forcément qu’une position de séquence génétique portant un A ait la même chance de devenir un T, G ou C, par exemple). On a donc un nombre de caractères bien supérieur à une approche morphologique, voire supérieur par de nombreux ordres de grandeur si l'on compare des génomes entiers. En effet, si les études phylogénétiques se sont longtemps basées sur un ou plusieurs gènes, la génétique moderne (dans le sillage du Human Genome Project/Projet Génome Humain de la fin du XXe siècle) est caractérisée par des campagnes de séquençage de génomes entiers d'espèces (voir l’article sur le projet DIVE-Sea). Ainsi, on a aujourd'hui des phylogénies basées sur des séquences de milliards de caractères.
Petit précis de méthodologie
Générer un arbre phylogénétique pour un échantillon d'organismes à partir de leurs caractères est un processus avec plusieurs étapes séquentielles. On cherche évidemment à générer le “meilleur arbre”, soit directement à l'assemblage initial, à l’aide de mesures de similarité (méthode généralement dépréciée), soit par recherche d’arbres optimaux - ceux qui requièrent le moins d'hypothèses, ou paraissent mathématiquement les plus plausibles. Comparer tous les arbres dans une recherche exhaustive est une démarche impossible qui prendrait un temps quasi infini avec tous sauf les échantillons les plus réduits. On doit donc chercher à comparer efficacement un grand nombre d'arbres. Le procédé général est ainsi le suivant : coder les caractères et/ou aligner les séquences ; générer un arbre initial de bonne qualité ; varier l'arbre à l'aide de permutations pendant un temps défini ; mesurer et comparer la qualité de chaque arbre généré ; générer un arbre de consensus (strict ou par majorité) si plusieurs arbres de qualité équivalente apparaissent. L'arbre généré par un tel processus est en fait un réseau sans sens donné, qui doit être “enraciné” grâce au placement d’un taxon dit « outgroup » déjà inclus dans l’échantillon (ce taxon est un parent proche du groupe d'organismes étudié, mais établi comme externe à ce groupe, par hypothèse de départ). On peut également donner un score de confiance aux branches de l’arbre final, à l’aide de méthodes de rééchantillonnage (boostrap ou jackknife).
La sélection des “meilleurs arbres” est la phase méthodologique phare. Trois méthodes prépondérantes existent. La méthode de parcimonie, la plus traditionnelle, surtout pour les données morphologiques, vise à minimiser le nombre de changements d’états de caractères présents dans l’arbre (et donc d’hypothèses nécessaires pour notre arbre - qui modélise un schéma d’évolution). Les méthodes statistiques, les plus largement utilisées et notamment en phylogénie moléculaire, sont la maximisation de vraisemblance (on estime la vraisemblance mathématique des arbres selon un modèle d’évolution) et la méthode d’inférence Bayésienne (on estime la probabilité postérieure d’un arbre selon un modèle et une probabilité antérieure donnée). Certaines méthodes font plus ou moins consensus chez les chercheurs, notamment l’inférence Bayésienne, mais elles peuvent varier en efficacité selon la nature d’un échantillon et les problèmes qu’il peut présenter.
Quelques problématiques
La phylogénie peut en effet rencontrer des phénomènes biologiques problématiques, pouvant affecter les résultats présents dans un arbre. Les échantillons avec des caractères morphologiques, en particulier ceux basés sur des fossiles, peuvent souffrir de données manquantes (états de certains caractères qu’on ne peut pas coder pour certains membres de l'échantillon). L'évolution convergente et parallèle (à la fois dans les données anatomiques et moléculaires, voir article à venir), qui constituent de l’homoplasie, par opposition à de l’homologie, peuvent générer de fausses synapomorphies apparentes dans un arbre. Le transfert horizontal de gènes et le phénomène d’hybridation mettent à mal la structure basique d’un arbre, qui exclut la reconnexion des branches une fois que des lignées sont établies. Dans le domaine moléculaire, les duplications de gènes ou de séquences de gènes sont communes. Si le gène B du saumon est présent chez l'humain et le cœlacanthe sous la forme des gènes B1 et B2, il devient plus difficile d’établir une comparaison directe. Il se produit même des dédoublements de génome entiers (polyploïdie), dont plusieurs sont théorisés dans l'histoire des poissons et des amphibiens, et qui sont communs au sein des plantes. Tous ces cas de figure au sein d'un échantillon peuvent introduire de la confusion au sein des arbres résultants.
L'usage de génomes entiers dans la phylogénie offre une très grande quantité de données, mais peut parfois ainsi dissimuler un grand nombre de loci génétiques potentiellement problématiques (un locus est une séquence génétique spécifique, un terme plus général que “gène”). En général, un arbre phylogénétique est d’autant mieux compris et soutenu que ses données sont bien étudiées. L’annotation détaillée d’un génome est un processus long et en amélioration constante, mais nécessaire au moins en partie pour une phylogénie bien menée. Les données moléculaires sont donc généralement moins bien comprises dans le détail par rapport aux données morphologiques, mais elles offrent l’avantage de générer des caractères naturels et moins subjectifs, puisqu’ils ne sont pas définis par les chercheurs eux-mêmes (et non potentiellement soumis à débat).
Des résultats probants
Nous savons ainsi d’où vient la phylogénie, comment elle fonctionne, et là où elle fonctionne moins bien. Mais où en est la discipline actuellement ? À l'heure des banques de données génomiques, la phylogénie moléculaire prend de plus en plus le pas sur la phylogénie morphologique. Celle-ci restera cependant toujours d’actualité, car nécessaire pour les espèces fossiles qui n’ont généralement pas de molécules à fournir, excepté quelques spécimens de moins de 1 million d’années, ou encore une séquence de collagène prise d’un os de Tyrannosaurus Rex pour être comparée à celle de crocodiles et d’oiseaux. Elle sert aussi crucialement à informer la génération de phylogénies moléculaires. Certains tentent une approche inclusive, mais quand une étude combine une dizaine de caractères morphologiques avec des milliards de molécules, ces données ne font pas long feu. Entretemps, l’apport de grandes études génétiques et génomiques a permis de faire des pas de géant dans notre connaissance de certains clades et leurs relations entre eux, comme les grandes familles d’oiseaux, ou les différents phylums d'animaux “invertébrés” (au sein desquels les vertébrés ne forment qu’un petit clade). Il reste encore de nombreux coins de l’arbre du vivant à élucider, et toujours plus de nouvelles données à intégrer. Peut-être de nouvelles méthodes pourront-elles amener de grandes avancées. Certains explorent l'usage de réseaux phylogénétiques plutôt que d’arbres stricts, prenant en compte l’ubiquité du partage génétique chez des clades comme les bactéries.
![A simplified phylogeny of birds according to Prum et al. [19] based on nucleotide sequences of 259 nuclear genes.](https://lh7-rt.googleusercontent.com/docsz/AD_4nXf1WbP98TT4c9HbPXf5Wx5YE_TOWQDpZLfNmAgPeXGqYp5blNicFUh3hofMqdgiW2QE2hdCGK4Jjz_D2j-6suuetXKLoiujdZ3Isty5Oko6HcXDz0p8CKLDiudCouankpT13Ol5Ckv_GKzYbpNy2OcNPIDYElIoG2YrqbBhr6OOnFtqE1F6?key=WtSWHcvAyzjeiubZvu3uwY7V)
Synthèse d’un arbre phylogénétique moléculaire des oiseaux, 2015.
Les arbres produits par les études phylogénétiques ne sont que des reconstitutions, et forment donc des hypothèses sur notre connaissance des liens qui unissent les espèces. Comme toute science, on n'espère pas élucider parfaitement l’arbre du vivant, ce qui serait impossible. On cherche à en faire, à l'aide des données et outils dont nous disposons, la meilleure reconstitution possible, et une reconstitution toujours plus juste.
Glossaire
Phylogénie: branche de la biologie qui étudie (et cherche donc à déterminer et cartographier) les liens de parenté entre êtres vivants. Elle peut être effectuée à l’échelle des individus (cela donne une généalogie), comme à l’échelle des populations, des espèces, ou d’autres taxons.
Arbre phylogénétique: (voir exemple-type ci-dessus) : diagramme en forme d’arbre représentant les liens de parenté entre les organismes. De manière plus scientifique, un arbre représente une hypothèse de schéma de parenté entre organismes, obtenu grâce à l’analyse de leurs caractères.
Taxon : catégorie ou groupe biologique établis sur la base de caractères partagés et/ou de descendance commune, utilisés dans la classification des organismes. Par exemple une espèce, un genre, une famille, un ordre, une classe ; mais également une sous-espèce ou une race domestique. Dans la taxonomie moderne, un véritable taxon doit être monophylétique.
Caractère : un élément d’un taxon que l’on peut diagnostiquer. Il peut être morphologique, moléculaire, comportemental… L’ensemble des caractères forme le phénotype d’un individu.
Cladistique : aussi appelée systématique phylogénétique, est une approche de la phylogénie se reposant sur l’identification de groupes monophylétiques, formant des taxons rigoureusement définis qu’on appelle clades. C’est la base de la pratique phylogénétique moderne.
Monophylétique : se dit d’un groupe biologique regroupant tous les organismes descendants d’un même ancêtre commun, sans exclusion. “Poisson”, au sens usuel du terme, n'est pas monophylétique, puisque les vertébrés terrestres descendent des poissons.
Homologie : similitude d’un caractère entre des organismes, sur la base d’une parenté commune (par opposition à une homoplasie, ou convergence de formes). Un caractère homologique propre à un groupe monophylétique est appelé synapomorphie.
Références
1)Asara, John M., et al. "Protein sequences from mastodon and Tyrannosaurus rex revealed by mass spectrometry." Science 316.5822 (2007): 280-285.
2)Darwin’s, Charles. "On the origin of species." published on 24 (1859): 1.
3)Dunn, Casey W., et al. "Broad phylogenomic sampling improves resolution of the animal tree of life." Nature 452.7188 (2008): 745-749.
4)Felsenstein, Joseph. "Inferring phylogenies." Inferring phylogenies. (2004) 664-664.
5)Hennig, Willi. Phylogenetic systematics. University of Illinois Press, 1999.*
6)Kitching, Ian J., Forey, Peter L., Humphries; Christopher J., Williams, David M. Cladistics: the theory and practice of parsimony analysis. No. 11. Oxford University Press, (1998)
7)Lee, Michael SY, and Alessandro Palci. "Morphological phylogenetics in the genomic age." Current Biology 25.19 (2015): R922-R929.
8) Prum, Richard O., et al. "A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing." Nature 526.7574 (2015): 569-573 . (synthèse dans image 3-CC 3.0)
9)Sibley, Charles G., Jon E. Ahlquist, and Burt L. Monroe Jr. "A classification of the living birds of the world based on DNA-DNA hybridization studies." The Auk 105.3 (1988): 409-423.
10)Wink, Michael. "DNA Analyses Have Revolutionized Studies on the Taxonomy and Evolution in Birds." Birds-Challenges and Opportunities for Business, Conservation and Research (2021): 3. (image 3-CC 3.0)
11) Zhao, Min, et al. "Exploring conflicts in whole genome phylogenetics: a case study within manakins (Aves: Pipridae)." Systematic biology 72.1 (2023): 161-178.
Références Images
Couverture: Haeckel, Ernst. The evolution of man. Vol. 1. D. Appleton, (1897)
Composantes Image 1 tirées de:
1)File:Comparison of size of orca and great white shark.svg - Wikimedia Commons - The Nature Box
2) File: Salmo salar.jpg - Wikimedia Commons -Timothy Klepp
3) File:Latimeria chalumnae replica.jpg - Wikimedia Commons -Citron
4) File:Male and female chicken sitting together.jpg - Wikimedia Commons - Andrei Niemimäki
5) Léonard de Vinci. L’ Homme de Vitruve. (vers 1492)
Images 2 et 3 tirées des sources 2), et 8)+10) respectivement
Commentaire ( 0 ) :


Partager
Catégories
Cela pourrait vous intéresser :
S'inscrire à notre newsletter
Nous publions du contenu régulièrement, restez à jour en vous abonnant à notre newsletter.